
If you had attended the American Association of Cancer Research (AACR) annual meeting this year (2019), you must have noticed a flurry of immunotherapy talks. This is the testament of the success achieved by cancer immunotherapy in recent years. From a couple of talks confined within a small room and the familiar crowd to the houseful sessions and Nobel prizes — immunotherapy has indeed come a long way. However, a lot is yet to be achieved if we wish to tame cancer using our immune system. The recent success of immunotherapy has come from immune checkpoint inhibitors and CAR-T cells. Both therapies rely on the activation of immune cells to wipe out the tumor. However, the success with checkpoint inhibitors and CAR-T is limited to a handful of patients and the rest still don’t benefit from immunotherapy. A lot of effort is now directed towards understanding why some tumors react to immunotherapy and others don’t? One of the determinants of immunotherapy is the presence of immune cells in the tumor: if there are a lot of immune cell infiltrations in the tumor then its called a “hot tumor”, likewise, if there are only a few immune cells in the tumor then its called “cold tumor”. Most of the tumors that react to checkpoint inhibitors are hot tumors, where activation of the immune cells (NK cells and T cells) leads to an antitumor response. However, in the case of a cold tumor, there aren’t enough immune cells to mount the antitumor response. Two of the recent article published in Cancer Discovery, (Sen et al. and Pantelidou and Sonzogni et al.) demonstrate how DNA damage response can be targeted to activate STING pathway-mediated immune cell recruitment.
What is STING and why is it gaining popularity in cancer immunotherapy circle?
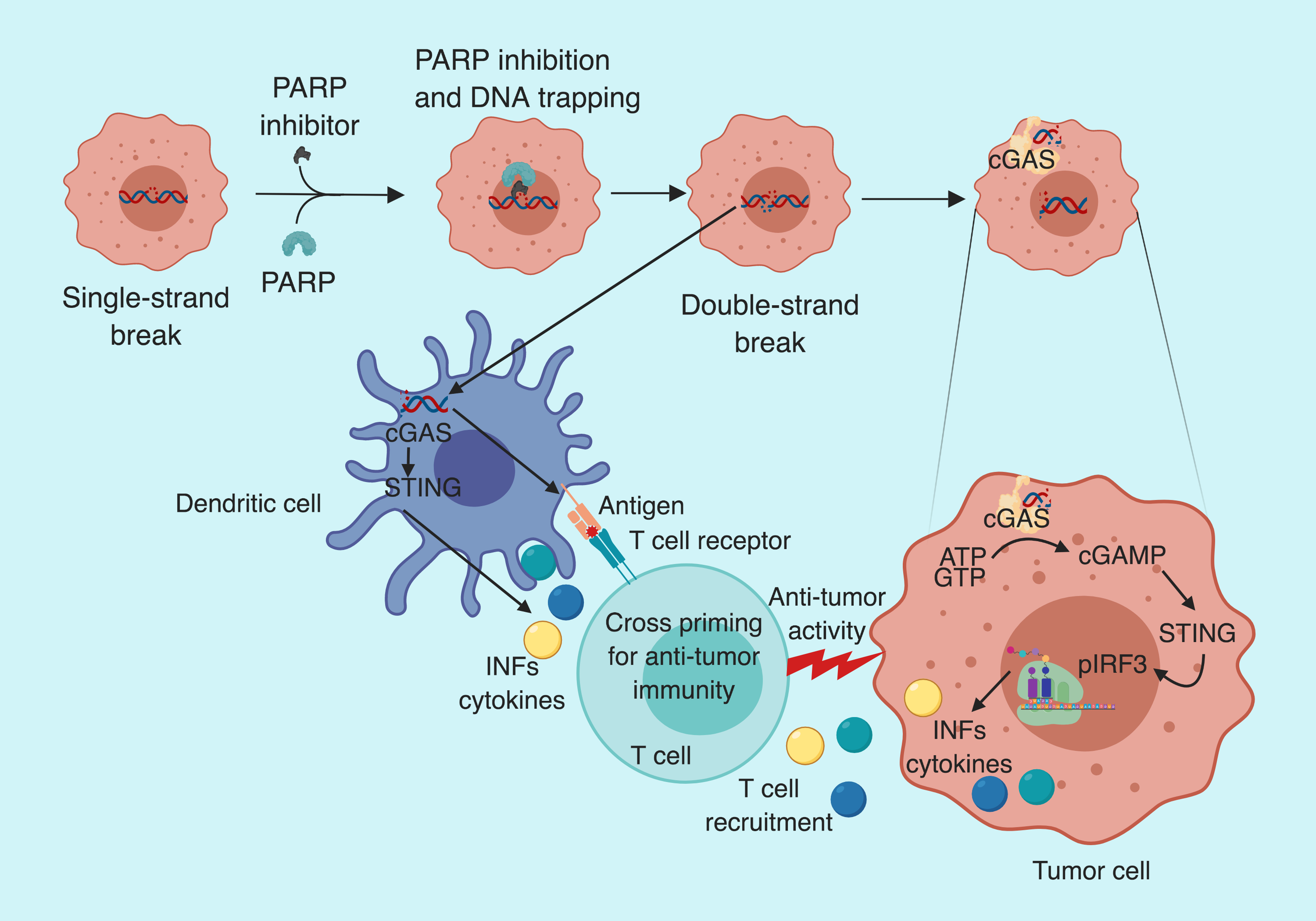
STING ( stimulator of interferon genes) is a key immune activator and the first line of response against infection. In Eukaryotes, the DNA is confined inside the nucleus unless there is mitochondrial or DNA damage. So, when the cells are infected with viruses, there DNA ends up in the cytoplasm. Since the DNA is not usually present in the cytoplasm, it is detected by the protein called cGAS (cyclic GMP-AMP synthase). cGAS is attached to the cell membrane by N-terminal phosphoinositide-binding domain and it functions as a sensor for cytosolic DNA. Once cGAS binds the DNA it converts ATP and GTP into a soluble secondary messenger, cyclic dinucleotide 2’3’-GMP-AMP (cGAMP). cGAMP binds to and activates STING pathway either in the same cell or in the neighboring cells. Activation of STING pathway leads to type I interferon production which is the warning signal for the immune system. Hence, attracting immune cells to take care of the inflammation.
Mechanism and status in Cancer?
Inflammation is a key player in fine-tuning the immune response. Acute inflammation recruits immune cells to the concerned site, whereas, chronic inflammation suppresses the immune system by upregulating checkpoint inhibitors. Activation of STING pathway triggers acute inflammation and leads to the recruitment of immune cells, an unfavorable condition for cancer cells. So to block the influx of tumor-infiltrating immune cells, cancer cells suppress STING pathway via genetic and epigenetic mechanisms. STING pathway activation has shown promising results in preclinical models, especially in combination with checkpoint inhibitors. However, systemic STING inhibition is non-tolerable and can lead to pathological conditions such as lupus. Hence, researchers have opted to inject STING agonist directly into the tumor to boost the inflammation. Although the results are promising, a more robust strategy to activate STING pathway, over onsite injection, would yield a better outcome.
In their recent publication, Pantelidou and Sonzogni et al. demonstrate how PARP inhibitors can be exploited to activate STING pathway in a tumor-specific manner in BRCA-deficient breast cancer models.
PARP inhibition in cancer
Poly (ADP-ribose) polymerase are the enzymes involved in DNA repair following single-strand breaks. PARP detects and binds to the region of single-strand damage via its zinc finger DNA-binding domain and recruits repair complex proteins. DNA repair is critical to cell survival and an unrepaired single-strand break (SSB) leads to double-strand break (DSB) which, if left unrepaired, will trigger cell death. Homologous recombination (HR) is considered the most precise DSB repair mechanism which is impaired in BRCA-mutant tumors. As expected, PARP inhibitors have shown promising results in BRCA-mutated tumors.
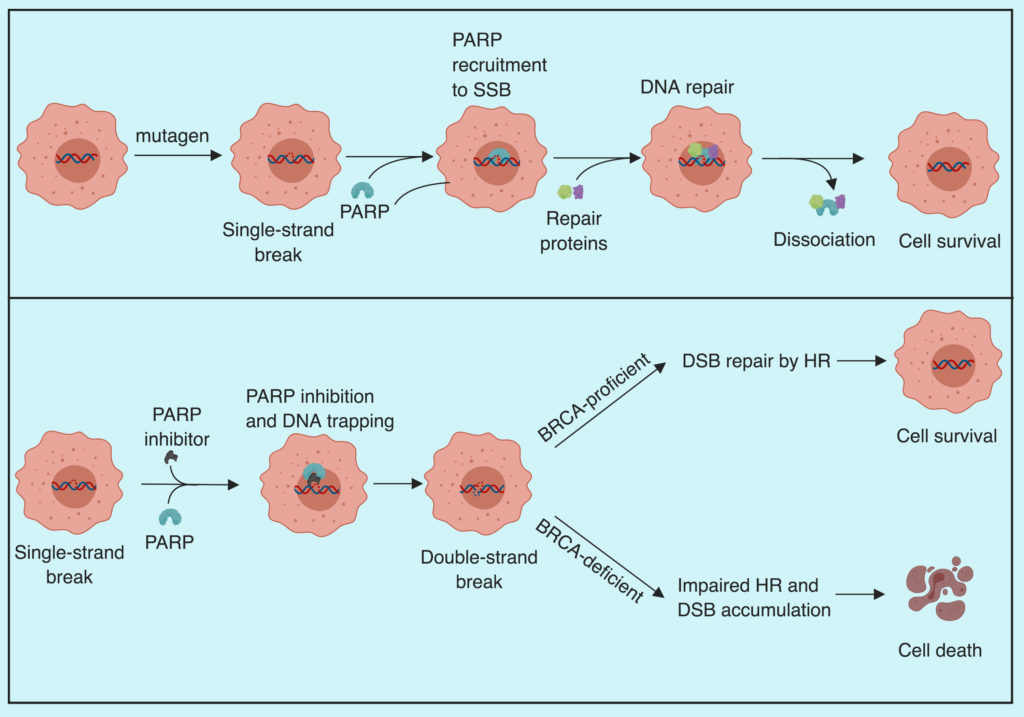
How does PARP inhibition work
In their recent work, Pantelidou and Sonzogni et al. demonstrate that the efficacy of PARP inhibitors depends on the recruitment of CD8+ T cells in BRCA mutant triple negative breast cancer (TNBC). To demonstrate this, they use genetically engineered mouse models (GEMM) of TNBC lacking functional BRCA and P53 proteins. To assess the possible role of PARP inhibitors in immune activation, the group transplanted the tumors from their GEMM into immunocompetent and immunodeficient mice and treated them with olaparib (PARP inhibitor). After the olaparib treatment, the median survival of immunocompetent mice was significantly higher compared to the immunodeficient mice (241 days/103 days). Suggesting the involvement of the immune system in tumor response to olaparib. To further pinpoint the role of immune cells, the group treated the tumors with CD8 neutralizing antibody along with olaparib and notice accelerated tumor growth and reduced median survival from 241 to 139 days. Suggesting the role of CD8+ T-cells in olaparib mediated tumor control. The group also noticed enrichment of CD3 and CD8 positive cells in treated samples and an increase in Granzyme B positive (activated) CD8 T-cells supporting the notion of CD8+ T-cells get activated after olaparib treatment. The group further noticed the reintroduction of BRCA1 in their GEMM led to the loss of olaparib mediated tumor control, validating the that loss of BRCA1 mediated DNA repair is crucial for PARP inhibitor anti-tumor efficacy.
PARP inhibitor (Olaparib) activates STING pathway both in tumor and dendritic cells
To explain the enrichment and activation of CD8+ T cell after olaparib treatment in BRCA deficient tumors, Pantelidou and Sonzogni et al. checked the STING pathway activation. They noticed an increase in cGAS-bound cytoplasmic micronuclei (indicative of cytoplasmic DNA); y-H2AX foci (indicative of DNA strand break); mRNA levels of INFβ, CCL5, and CCL10 (indicative of increased proinflammatory cytokine production); pTBK1 and pIRF3 (indicative of STING/TBK1/IRF3 pathway activation) in BRCA1 deficient murine tumor cells. Suggesting, olaparib mediated DNA damage leads to STING pathway activation and type I interferon-mediated proinflammatory chemokine production, which is known to recruit CD8+ T cells.
PARP inhibitor activates STING in tumor cells
The group also noticed an increase in pTKB and pIRF3 positive dendritic cells (DCs) after olaparib treatment. On further analysis, they observed a significant increase in DCs expressing major histocompatibility complex class II— indicating maturation and antigen presenting ability of DCs. However, treatment of cultured DCs with olaparib failed to activate STING pathway. Suggesting, that olaparib activates STING pathway in BRCA deficient tumor cells, which activates STING in DCs.
PARP inhibitors anti-tumor effect if STING dependent
To confirm the role of STING pathway in olaparib-mediated tumor control, Pantelidou and Sonzogni et al. knocked-out STING using CRISPR/Cas9 system in K14 cells derived from BRCA GEMM. Knocking out STING abolished Olaparib-dependent TBK1 phosphorylation and INFβ, CCL5, and CCL10 production. Furthermore, repletion of STING in STING KO cells led to olaparib-dependent TBK1 phosphorylation and induction of type I interferon-mediated chemokine production. The group then depleted cGAS and observed similar outcomes as with the STING knock out. Thus, suggesting that cGAS/STING pathways are required for PARP inhibitor-induced proinflammatory signaling.
STING activation is required for T cell recruitment
To assess the functional significance of STING pathway activation following PARP inhibition, the group analyzed T-cells status in STING KO mouse tumors after Olaparib treatment. They observed a decrease in CD8+ T-cells infiltration, granzyme B positive CD8 T-cells and pIRF3+ DC cells in STING KO mice. Furthermore, tumor burden in response to olaparib was significantly higher in STING KO mice compared to control. Suggesting the role of STING pathway in PARP inhibitor-mediated CD8+ T-cells recruitment and anti-tumor efficacy.
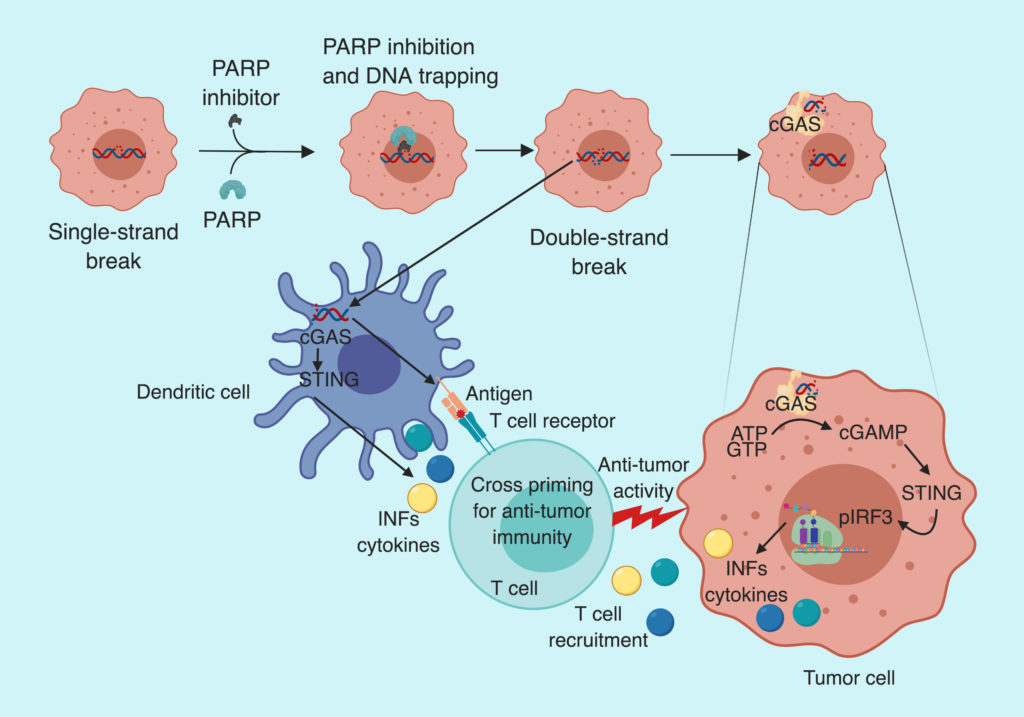
To summarize, Pantelidou and Sonzogni et al. demonstrate that the anti-tumor effect of PARP inhibitors is mediated via activation of cGAS/STING pathway, which leads to type I interferon-dependent cytokine production and recruitment of CD8+ T-cells in BRCA deficient tumors.
By Shishir Pant
Reference