
In the absence of functional apoptotic pathway, most chemotherapy and probably many other anticancer drugs suppress tumor via senescence. However, induction of senescence isn’t considered a good therapeutic outcome and many researchers are looking to combine cytostatic drugs, with other drugs which would selectively kill the senescent cells (
So, what is senescence?
Lately, senescence has stirred quite a few controversies among researchers due to the absence of a clear definition. Hayflick and Moorhead coined the term “cell senescence” in 1961 based on their observation that normal human fibroblasts (a type of cells) were unable to proliferate (grow) indefinitely in vitro (outside the body). Later on, it was shown that these cells were alive and their growth capacity was dependent on the donor age, linking senescence to aging. Epstein et al. in 1966 observed premature senescence in patients with Werner’s syndrome, accelerated aging disease, establishing the link between senescence and aging. So, for a long time, senescence has been considered as a permanent condition “an irreversible cell cycle arrest”. However, in recent years this dogma has been challenged by many researchers and the definition is shifting towards “stable cell cycle arrest”.
Molecular features of senescent cells
Although initially perceived to be an inactive state, senescent cells demonstrate a host of traits including secretion of growth factors, cytokines (ILs, MCP-2), cell surface molecules (ICAM, uPAR), pro-collagenase, and matrix remodeling enzymes – collectively known as senescence-associated secretory phenotype (SASP). Although the role of SASP is not very well defined, several recent publications imply that SASP is associated with active self-elimination of senescent cells via the immune system. Cytokines released by senescent cells attract the immune cells, which recognize their target via cell surface ligands such as ICAM. However, in the case of an impaired immune system, which is the hallmark of cancer, these senescent cells persist long-term in the body, resulting in a pathological pro-inflammatory condition which supports tumor growth.
So, why are we interested in senescence for cancer therapy?
In the recent article published in the journal Science, Ruscetti et al. show that the combination of MEK and CDK4/6 inhibitor drives tumor cells towards senescence which are later cleared by SASP mediated recruitment of NK cells (a type of immune cell).
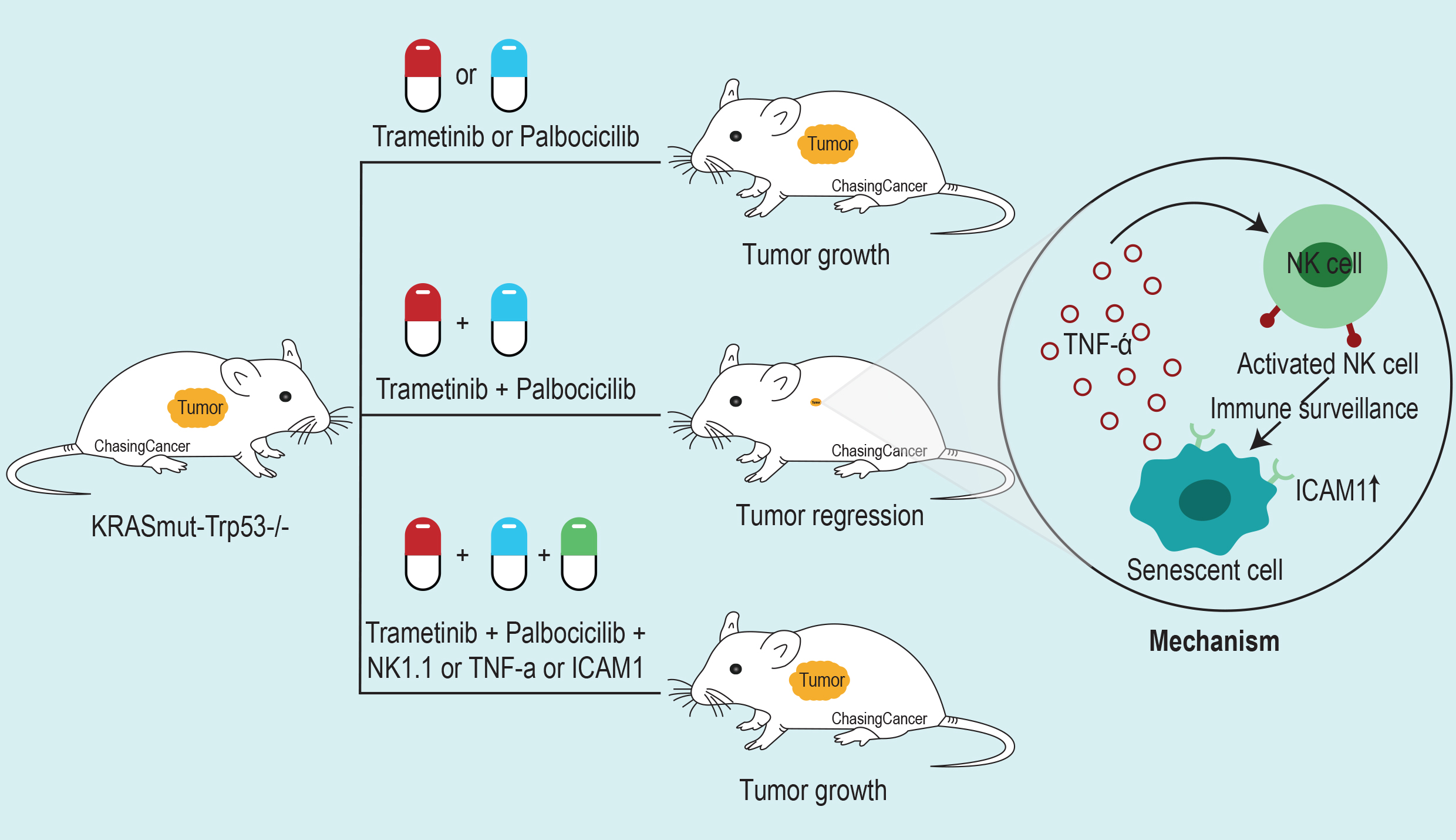
To show that NK cells are essential for tumor regression by MEK and CDK4/6 inhibitor combination the group first tested the efficacy of MEK inhibitor (Trametinib) with CDK4/6 inhibitors (Palbociclib, Ribocicilib, and Abemacicilib) in colony formation assay using KRAS-mutant lung and pancreatic cancer cell lines. They show that the combination was able to inhibit the proliferation of KRAS-mutant cell lines and also noticed reduced levels of phospho-retinoblastoma (RB) and phosphor-ERK proteins, which are the targets of CDK4/6 and MEK respectively

To test the efficacy of the combination in vivo
Ruscetti et al. used two KRAS-mutant lung cancer patient-derived xenografts (PDXs) where they observed the reduction in tumor growth with the Trametinib and Palbociclib combination at low and high doses. However, human xenograft experiments were done in immune-compromised mice so to assess the role of the immune system associated with this drug combination the group used a syngenic KRAS mutant (KrasG12D/+); Trp53 negative (P53-/-) mouse model. They observed a reduction in tumor growth with the combination in immunocompetent KP mice which was lost when the same cells were transplanted to immune-deficient mice. Suggesting the possible role of the immune system associated with the anti-tumor outcomes of this drug combination. On subsequent analysis of immune cells using KP mouse model, Ruscetti et al. found an increase in activated NK cell in the combination treated tumor samples indicated by the presence of CD107a cell surface marker and a decrease in myeloid-derived suppressor cells (MDSCs). To further pinpoint the immune components responsible for the anti-tumor effect, the group depleted/blocked macrophages, T cells, NK cells and also combined their drug with anti PD1 inhibitors. Strikingly, only the blockade of NK cell was able to abolish the effect of the combination treatment.
So, how does MEK and CDK4/6 inhibitors induced NK cell activation?
MEK and CDK4/6 inhibition lead to dephosphorylation of Rb (Retinoblastoma) protein which then inhibits cell proliferation and initiates cellular senescence. Ruscetti et al. looked into the senescence markers in KRAS-mutant lung cancer cell lines and found increased SA-beta-gal positive cells, upregulation of SASP associated gene expression, and stable cell-cycle arrest. All of which are the markers of senescent cells. The group further shows that NK cell blockade leads to accumulation of SA-beta-gal positive cells in KP transplant lung tumors and an increased tumor burden. Highlighting, the role of NK cells in senescent cells clearance.
To identify the molecular regulators of NK cell-mediated tumor control,

Ruscetti et al. looked into the role of NF-κB. Since NF-κB is a known regulator of SASP associated gene expression, the group then knock-down p65 subunit of NF-κB. While the cells still undergo senescence, there was a decrease in SASP associated gene expression, and cell death with the combination therapy. Implying that the activation of NK cell might be regulated via NF-κB mediated SASP gene expression.
They further blocked the activity of several SASP related factors and found that blockade of TNF-α activity in KP mouse model was able to prevent tumor NK cell-associated tumor killing when treated with the combination therapy. Thus, showing that the TNF-α gene expression, related
The group further used an autochthonous KRAS-mutant lung cancer model to study the influence of NK cell immune surveillance in a more physiologically relevant setting. Like in the previous model, they observed tumor regression with the combination therapy which was blunted in the presence of NK cell, ICAM-1, and TNF-α blocking antibody.
Overall,
Ruscetti et al. demonstrate that senescence inducing therapy can lead to tumor regression and extend survival in presence of functional NK cell-mediated immune surveillance.
by Shishir Pant
Reference
Ruscetti, M., et al. (2018). “NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination.” Science 362(6421): 1416-1422.